K-mer Spectral Error Trimming¶
(Optional)
khmer documentation: http://khmer.readthedocs.io/en/latest
If you plot a k-mer abundance histogram of the samples, you’ll notice something: there’s an awful lot of unique (abundance=1) k-mers. These are erroneous k-mers caused by sequencing errors.
In a new Python3 Jupyter Notebook, run:
cd /mnt/work
and then
!abundance-dist-single.py -M 1e9 -k 21 SRR1976948_1.fastq.gz SRR1976948_1.fastq.gz.dist
and in another cell:
%matplotlib inline
import numpy
from pylab import *
dist1 = numpy.loadtxt('SRR1976948_1.fastq.gz.dist', skiprows=1, delimiter=',')
plot(dist1[:,0], dist1[:,1])
axis(xmax=50)
Many of these errors remain even after you do the Trimmomatic run; you can see this with:
!abundance-dist-single.py -M 1e9 -k 21 SRR1976948_1.qc.fq.gz SRR1976948_1.qc.fq.gz.dist
and then plot:
dist2 = numpy.loadtxt('SRR1976948_1.qc.fq.gz.dist', skiprows=1, delimiter=',')
plot(dist1[:,0], dist1[:,1], label='untrimmed')
plot(dist2[:,0], dist2[:,1], label='trimmed')
legend(loc='upper right')
axis(xmax=50)
This is for two reasons:
First, Trimmomatic trims based solely on the quality score, which is a statistical statement about the correctness of a base - a Q score of 30 means that, of 1000 bases with that Q score, 1 of those bases will be wrong. So, a base can have a high Q score and still be wrong! (and many bases will have a low Q score and still be correct)
Second, we trimmed very lightly - only bases that had a very low quality were removed. This was intentional because with assembly, you want to retain as much coverage as possible, and the assembler will generally figure out what the “correct” base is from the coverage.
An alternative to trimming based on the quality scores is to trim based on k-mer abundance - this is known as k-mer spectral error trimming. K-mer spectral error trimming always beats quality score trimming in terms of eliminating errors; e.g. look at this table from Zhang et al., 2014:

The basic logic is this: if you see low abundance k-mers in a high coverage data set, those k-mers are almost certainly the result of errors. (Caveat: strain variation could also create them.)
In metagenomic data sets we do have the problem that we may have very low and very high coverage data. So we don’t necessarily want to get rid of all low-abundance k-mers, because they may represent truly low abundance (but useful) data.
As part of the khmer project in my lab, we have developed an approach that sorts reads into high abundance and low abundance reads, and only error trims the high abundance reads.
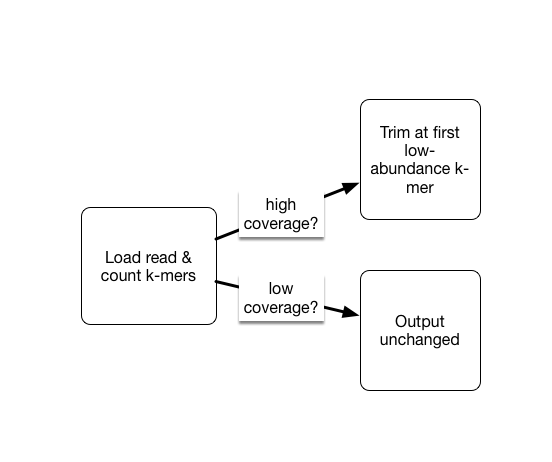
This does mean that many errors may get left in the data set, because we have no way of figuring out if they are errors or simply low coverage, but that’s OK (and you can always trim them off if you really care).
To run such error trimming, use the command trim-low-abund.py
(at the command line, or prefix with a ‘!’ in the notebook):
interleave-reads.py SRR1976948_1.qc.fq.gz SRR1976948_2.qc.fq.gz |
trim-low-abund.py -V -M 8e9 -C 3 -Z 10 - -o SRR1976948.trim.fq
Why (or why not) do k-mer trimming?¶
If you can assemble your data set without k-mer trimming, there’s no reason to do it. The reason we’re error trimming here is to speed up the assembler (by removing data) and to decrease the memory requirements of the assembler (by removing a number of k-mers).
To see how many k-mers we removed, you can examine the distribution as above,
or use the unique-kmers.py script:
unique-kmers.py SRR1976948_1.qc.fq.gz SRR1976948_2.qc.fq.gz
unique-kmers.py SRR1976948.trim.fq
LICENSE: This documentation and all textual/graphic site content is licensed under the Creative Commons - 0 License (CC0) -- fork @ github. Presentations (PPT/PDF) and PDFs are the property of their respective owners and are under the terms indicated within the presentation.